Arthritis is a major health problem in the world, and nearly 50 million people in the United States alone have the disease (see http://www.arthritis.org/). Arthritis causes more disability than any other chronic inflammatory disease, and the Center for Disease Control reported that the financial costs attributable to arthritis and other rheumatic conditions in the United States in 2003 alone was ~ $130 billion which equaled 1.2% of the country’s gross domestic product that year (see http://www.cdc.gov/arthritis/data_statistics/cost.htm).
Fibroblast-like synoviocytes, MCs, and other immune cells markedly increase in number in the diseased synovium of patients with rheumatoid arthritis (RA). This inflammatory state leads to proteolytic loss of aggrecan proteoglycans and other prominent constituents of cartilage’s extracellular matrix. This is followed by bone loss and joint remodeling. MCs are present in increased numbers at the sites of cartilage erosion in the joints of patients with RA; they often comprise up to 5% of the immune cells in synovial tissue. Histamine and immunoreactive hTryptase-β have been detected in the synovial fluids of patients with arthritis. While circumstantial evidence was obtained in the 1980s that activated synovial MCs contribute to RA, the cell’s contribution to joint inflammation and cartilage loss remains poorly understood in humans. MC‑restricted tryptase•heparin complexes have pro‑inflammatory activity, and the mouse tetramer-forming tryptases mMCP-6 and mMCP-7 are abundant in the synovium of arthritic WT BALB/c mice. While MCs have been implicated in RA and some animal models of the disease, no direct evidence for a MC-restricted tryptase in the pathogenesis of inflammatory arthritis had been shown before our studies.
It is now known that activated mouse MCs release >50 biologically active factors, many of which have contrasting bioactivities. For example, some populations of MCs activated via their high-affinity IgE receptors produce the arthritogenic cytokines interleukin (IL)-1β and tumor necrosis factor-α but also the anti-arthritogenic cytokine IL-13. Thus, a major problem using MC‑deficient mouse strains in a disease model is that it is difficult to deduce what is going on if one prevents the expression of all mediators from a MC by knocking out all of these effector cells in a tissue site like the synovium. We therefore created transgenic mice that lack heparin (Humphries et al., Nature 1999;400:769) (view Humphries et al.) and different combinations of mMCP‑6 and mMCP‑7 (view Thakurdas et al) to evaluate in a more definitive manner the contributions of MC-restricted tryptase•heparin complexes in the widely used methylated bovine serum albumin (mBSA)/IL‑1β (McNeil et al., Arthritis Rheum. 2008;58:2338) (view McNeil et al.) and K/BxN mouse serum‑transfer arthritis models (Shin et al., J. Immunol. 2009;1982;647) (view Shin et al.).
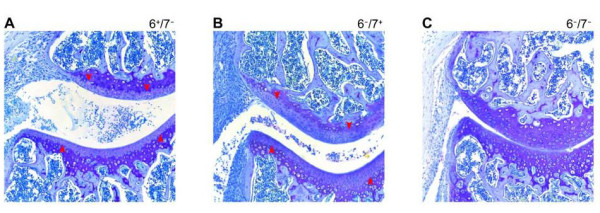
Fig. 6. Experimental arthritis is dependent on the tetramer-forming tryptases mMCP-6 and mMCP-7. Experimental arthritis was induced in B6 mice that lacked mMCP-7 (A), mMCP-6 (B), or both tetramer-forming tryptases (C). Tissue sections from the resulting three groups of animals were stained with toluidine blue to evaluate the loss of the aggrecan proteoglycans in the diseased cartilage. Only those transgenic mice that lacked both MC tryptases failed to lose their aggrecan proteoglycans, as documented by decreased histochemistry (red arrowheads). Similar data were obtained when cartilage biopsies were stained with anti-aggrecan antibodies (data not shown) (from McNeil et al., Arthritis Rheum. 2008;58:2338).
Arthritis was induced in the knee joints of mBSA/IL‑1β-treated mMCP‑6+/mMCP‑7‑ and mMCP‑6‑/mMCP‑7+ B6 mice and mMCP‑6+/mMCP‑7+ BALB/c mice, and numerous activated MCs that had exocytosed the contents of their secretory granules were observed in the joints of the diseased animals. In contrast, arthritis was markedly reduced in heparin-deficient mice and in mMCP‑6‑/mMCP‑7‑ transgenic mice (see Figure 6). Thus, tryptase•heparin complexes play important roles in mBSA/IL‑1β-induced arthritis. Because mMCP‑6 and mMCP‑7 can compensate for each other in this arthritis model, the elimination of both tryptases is necessary to uncover the prominent roles of these serine proteases in joint inflammation and destruction.
The importance of the two MC-restricted tetramer-forming tryptases in K/BxN arthritis was also evaluated . While mMCP-6 was more important than mMCP-7 in this disease model, the data in the Shin et al. study agreed with the data from the McNeil et al. study. In this second experimental model, arthritis also was dependent on the chemokine receptor CXCR2 which regulates neutrophil chemotaxis. In support of these data, exposure of human and mouse fibroblast-like synoviocytes to hTryptase‑β•heparin or mMCP‑6•heparin complexes resulted in expression of numerous neutrophil-responsive chemokines that bind to CXCR2.
Our proteomics, histochemistry, and immunohistochemistry data revealed substantial loss of cartilage-derived aggrecan proteoglycans in the arthritic joints of WT mice but not tryptase-deficient mice in both arthritis models. Aggrecan is a major constituent of cartilage, and it gives this tissue its intrinsic weight-bearing properties. The G1 and G2 globular domains of aggrecan reside at the protein’s N terminus. The G1 domain physically immobilizes aggrecan in the cartilage matrix by binding to hyaluronan. The amino acid sequence downstream of the G2 domain is the part of the proteoglycan that is substituted with glycosaminoglycans that attract water to confer the osmotic swelling pressure that enables cartilage to resist compressive loads. Aggrecan degradation (termed aggrecanolysis) is a key feature of arthritis, yielding fragments of this proteoglycan that are readily detected in the patient’s cartilage and synovial fluids. The most detrimental aggrecanolysis occurs within the interglobular domain that separates the G1 and G2 domains. It is mediated, in part, by MMPs. The mechanism by which latent MMPs are activated in cartilage is unknown. However, MMP-3 and MMP-13 are known to cleave aggrecan at its DIPEN↓FFGVG site.
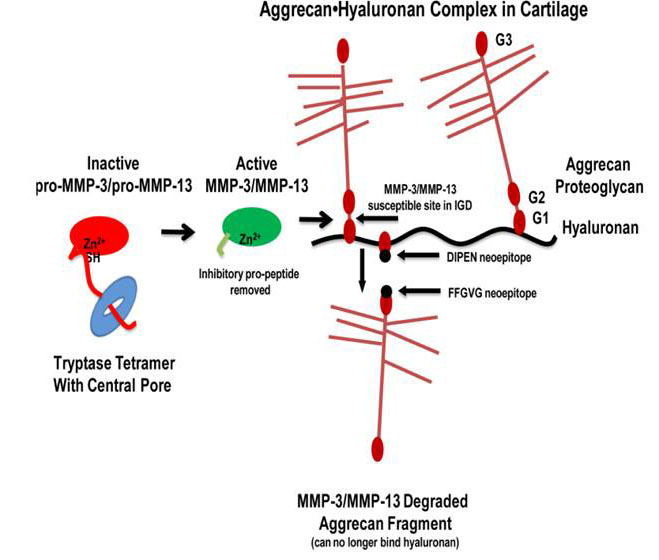
Fig. 7. Tryptase-dependent aggrecanolysis. As depicted in this sematic model, once the MC tryptases in the inflamed synovium proteolytically remove the propeptides of the inactive MMP‑3 and MMP-13 zymogens, the resulting enzymatically active MMPs cleave aggrecan proteoglycan between its G1 and G2 domains. This results in fragments cannot be retained in the diseased cartilage (from Magarinos et al., J. Immunol. 2013;191:1404).
The McNeil et al. 2008 and Shin et al. 2009 studies revealed that our tryptase-null mice contained fewer neutrophils in their synovium and reduced loss of aggrecan proteoglycans from the extracellular matrix of their articular cartilage during inflammatory arthritis relative to diseased WT mice. While these data could have been a consequence of fewer neutrophils in the arthritic joints, we noted in 1992 (Stevens et al., Arthritis Rheum. 1992;35:325) (view Stevens et al.) that an undefined serine proteinase present in rat peritoneal MCs could cleave the N terminus of aggrecan in vitro in the absence of neutrophils, thereby resulting in fragments of aggrecan that could not recognize hyaluronan. In 2013 (Magarinos et al., J. Immunol. 2013;191:1404) (view Magarinos et al.), we used ex vivo mouse femoral head explants to determine how mMCP-6 and hTryptase-β mediate aggrecanolysis in the absence of neutrophils. Exposure of the explants ex vivo to recombinant hTryptase-β, recombinant mMCP‑6, or lysates of WT mouse peritoneal MCs significantly increased the levels of enzymatically active MMPs and significantly induced aggrecan loss into the conditioned media relative to replicate explants exposed to medium alone or lysates of mMCP‑6-null peritoneal MCs. Treatment of cartilage explants with tetramer-forming tryptases generated aggrecan fragments containing a C-terminal DIPEN neoepitope, consistent with MMP-dependent aggrecanolysis. The abilities of mMCP-6-containing lysates from WT peritoneal MCs to induce aggrecanolysis were prevented by inhibitors of MMP‑3 and MMP‑13. Finally, recombinant hTryptase-β was able to activate latent pro‑MMP-3 and pro‑MMP‑13 in vitro. The accumulated data suggest that human and mouse tetramer-forming tryptases are convertases that mediate cartilage damage in arthritis, in part, by activating the zymogen forms of MMP-3 and MMP-13 (see Figure 7). In summary, the MC’s tetramer-forming tryptases participate in experimental arthritis by at least two mechanisms, namely by inducing the accumulation of neutrophils in the diseased joint and then by proteolytically activating constitutively expressed MMP zymogens.
We showed that the expression and granule accumulation of the tryptase mMCP-6 in MCs is highly dependent on the cytokine IL-33 and its receptor IL1RL1 (Kaieda et al., J. Biol. Chem. 2010;285:21478) (view Kaieda et al.). In support of the Stevens et al. 1992, McNeil et al. 2008, Shin et al. 2009, and Magarinos et al. 2013 studies, others implicated IL-33 and IL1RL1 in arthritis in mice and humans. The accumulated data raises the possibility that inhibitors of hTryptase-β might have efficacy in the treatment of humans with arthritis.
Human chymase-1 (hCMA1) is a serine protease stored in the secretory granules of the constitutive MCs in most connective at a 1:1 molar ration with human MC carboxypeptidase A [hMC‑CPA; also known as carboxypeptidase A3]. The mouse ortholog of hCMA1 is mouse MC protease-5 (mMCP-5). Using a homologous recombination approach, a C57BL/6 mouse line was recently created that possessed a disrupted mMCP-5 gene. We show in a submitted study that these transgenic mice can be used to better understand the function of mMCP-5 (and by inference hCMA-1). In this regard, we show mMCP-5 is essential for packaging of mMC-CPA protein in the MC’s granule. We then show that fibronectin is a target of mMCP-5, that this serine protease regulates matrix metalloproteinase (MMP) -9/gelatinase B expression in vivo, and that mMCP-5 has prominent adverse roles in two widely used arthritis models. Whatever the mechanism by which mMCP-5 participates in arthritis, transgenic mice that lack this hCMA1 ortholog are viable and have no developmental abnormalities. Moreover, loss of mMCP-5 does not negatively impact the expansion and phenotype of the T-cell dependent MCs in the animal’s jejunum. Importantly, the MCs in our mMCP-5-null mice continue to express mMCP-6 (the mouse ortholog of hTryptase‑β) which is a tryptase that has beneficial roles in innate immunity, acquired immunity, and the internal accumulation of prevention of fibrin-platelet clots. In terms of the human relevance of our mMCP-5 data, anti‑hCMA1 therapy might be highly effective in the treatment of those with arthritis without causing serious adverse side effects.
Comments are closed.